
Application and Impact of QSAR on Drug Discovery Awarded ECACS Bronze Award
Quantitative Structure-Activity Relationship (QSAR) modeling is computational modeling software used in virtual drug discovery. It is commonly used to predict the biological activity of chemical compounds based on their molecular structure and to quantify the data, enabling it to be used in other machine learning software. QSAR has become a central component of modern drug discovery pipelines, particularly in early-stage screening of developmental drugs. However, the accuracy and reliability of QSAR models are constrained by several key factors, including the quality and consistency of input data, the selection of appropriate molecular descriptors, and the use of underdeveloped validation methods. Additionally, issues such as overfitting and limited applicability domains can reduce a model’s ability to make predictions for new compounds or apply its predictions to more general test sets. This literature review examines how QSAR is applied in virtual drug discovery and evaluates the primary factors that influence its predictive performance.
Introduction
Quantitative Structure-Activity Relationship (QSAR) modeling is a computational approach used in virtual drug discovery. It is commonly used to predict the biological activity of chemical compounds based on their molecular structure and to quantify the data, enabling it to be used in other machine learning software. QSAR has become a central component of modern drug discovery pipelines, particularly in early-stage screening of developmental drugs. However, the accuracy and reliability of QSAR models are constrained by several key factors, including the quality and consistency of input data, the selection of appropriate molecular descriptors, and the use of underdeveloped validation methods. Additionally, issues such as overfitting and limited applicability domains can reduce a model’s ability to make predictions for new compounds or apply its predictions to more general test sets. This literature review examines how QSAR is applied in virtual drug discovery and evaluates the primary factors that influence its predictive performance.
Foundation of QSAR
QSAR operates by interpreting a molecule’s structural properties into values called molecular descriptors. It then uses those values as input data for a predictive model. These descriptors can represent chemical information such as molecular weight, polarity, hydrophobicity, electronic properties, or surface area. The model learns statistical relationships between descriptors and a measured outcome, commonly biological activity, accelerating drug discovery by predicting how new compounds will behave.
The many different types of QSAR models are categorized by their structural dimensionality. Two-dimensional (2D) QSAR uses flat and graph-based representations of molecules. It records connectivity without accounting for 3D geometry. Three-dimensional (3D) QSAR builds on this model, incorporating the spatial arrangement of atoms and accounting for how a molecule’s shape influences its interactions and function with a biological target. Four-dimensional (4D) is even more complex, incorporating multiple orientations to capture the flexibility of molecules in a solution. Each progressive dimension increases descriptive power but at the cost of greater data requirements.
QSAR applications are practical across a multitude of scientific domains. In drug discovery, it is used to monitor and check large sets for candidates likely to exhibit therapeutic activity, reducing the number of compounds that need to be synthesized and tested in the lab. In toxicology, QSAR models predict whether a compound poses environmental or health hazards. This supports regulatory assessments by eliminating the use of animal testing. In environmental science, QSAR has been used to assess the risks posed by pharmaceuticals entering ecosystems through manufacturing emissions, human excretion, and improper disposal. Researchers call these environmentally persistent pharmaceutical pollutants (EPPPs). These compounds, engineered to resist metabolic degradation, can accumulate in aquatic systems and harm wildlife in documented ways. In a scientific study on the effects of EPPPs on ecosystems, natural and synthetic estrogens have been linked to feminizing effects in fish and amphibians, while drug residues have been connected to disrupted behavior in aquatic species. QSAR assesses risks across thousands of compounds that would be far too expensive to test individually.
Evolution of QSAR
Over the years, QSAR has changed from using simple mathematical formulas to applying artificial intelligence to make much more advanced predictions. Early QSAR models focused mainly on identifying direct connections between molecular structure and biological interactions through basic statistical analysis. These initial models were useful for understanding smaller datasets but struggled with the complexity of real biological systems. As computing technology continued to evolve, researchers developed more advanced methods capable of processing larger volumes of data and recognizing more complex patterns. This transition marked an important shift in QSAR from a rudimentary statistical model to a predictive, data-driven tool that has had a significant impact on the pharmaceutical industry.
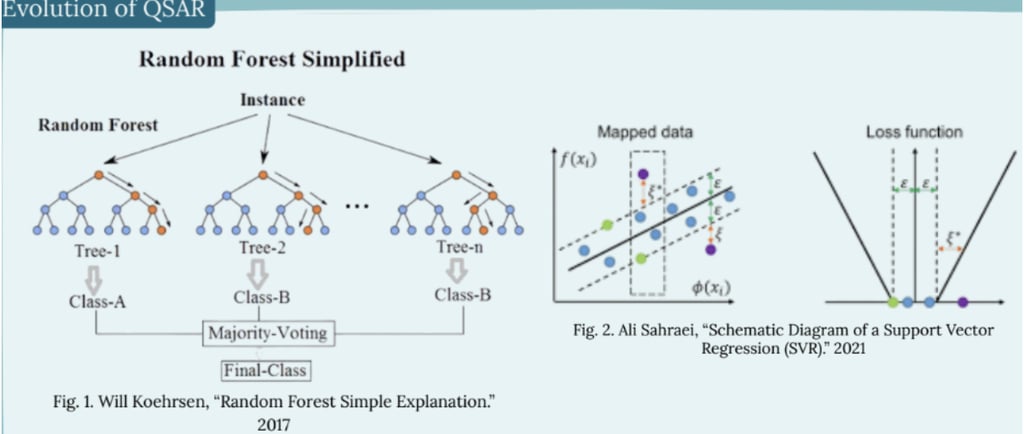
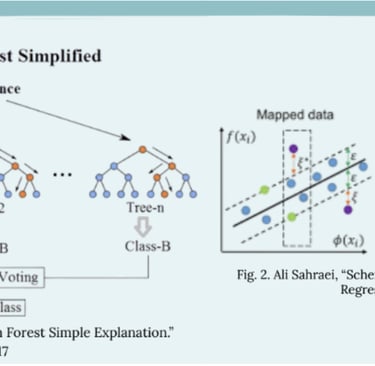
Traditional QSAR methods commonly use techniques such as Multiple Linear Regression (MLR), Partial Least Squares (PLS), and k-Nearest Neighbors (k-NN). These approaches relied much more heavily on molecular descriptors, using chemical properties to provide a rough estimate of how chemicals might interact. This design was much simpler, allowing researchers to easily interpret relationships. However, their low predictive accuracy and difficulty with complex datasets made them inefficient for practical use. Machine learning QSAR has introduced much more advanced computational methods capable of overcoming the limitations of earlier models. Techniques such as Random Forests (Fig. 1) and Support Vector Machines (Fig. 2) enabled researchers to model nonlinear relationships that traditional methods could not capture. These improvements allowed systems to work with much larger datasets and more diverse molecular descriptors, improving reliability in virtual screening and drug discovery. Although these newer models greatly improved predictive capabilities, they still have limitations that are currently being addressed.
Deep Learning
Deep learning has significantly expanded the capabilities of QSAR by allowing models to learn directly from molecular structures and recognize much more complex patterns. Unlike earlier QSAR methods that relied heavily on manually selected descriptors, deep learning systems can automatically identify important molecular features from large datasets. Common deep learning methods used in QSAR include neural networks, graph neural networks (GNNs), graph isomorphism networks (GINs), SMILES-based transformers, and generative or reinforcement learning models. These systems are able to process chemical structures in much more advanced ways, especially by representing molecules as connected graphs of atoms and bonds. This allows deep learning QSAR models to achieve much higher predictive accuracy and work more effectively with complex biological systems and molecular interactions.
Deep learning has also allowed QSAR to move beyond simply predicting activity and into actually generating new compounds. Generative AI and reinforcement learning models can design entirely new molecules with specific desired properties, helping speed up the early stages of drug discovery. One major example of this was Insilico Medicine, which used AI-driven drug discovery systems to develop an anti-fibrotic drug candidate for idiopathic pulmonary fibrosis (IPF). According to reports published by Nature Biotechnology and Nature, the company was able to move from target discovery to a preclinical drug candidate in around 18 months, with the AI-designed drug entering Phase I clinical trials in under 30 months. This process is significantly faster than traditional pharmaceutical development timelines, which often take several years before reaching clinical testing. These advancements show how deep learning is helping make drug discovery faster, more scalable, and more efficient than earlier QSAR methods.
Limitations of QSAR
Despite many advances and practical applications of QSAR, there are still many limitations to its reliability. QSAR heavily depends on the quality of the data it is given to produce its final output. Too many descriptors cause overfitting, while too few lead to weak predictions. In addition to these challenges, there is a lack of standardization across studies. There are no consistent datasets or evaluation frameworks, leading to differences in descriptor sets and other factors in published models, as well as making comparison difficult. The QSAR DataBank (QDB) is a solution that aims to solve this problem. QDB is a system designed to store, standardize, and share QSAR model equations. It uses a standardized data format and a consistent structure for input data. It even holds all the data, including chemical structures, validation data, and model equations, allowing everyone to rebuild and verify the model. This also prevents “black box” QSAR, where accurate results are provided without showing the process and algorithms. QDB addresses the poor standardization and reliability of models by being fully transparent with data sets and algorithms.
The bridge between model complexity and human interpretation remains one of the most critical challenges in AI. QSAR is still mainly a “black box” algorithm, resulting in limited trust and interpretability by chemists. The model tells chemists which molecules are predicted to be active, but its “black box” approach leaves them without the underlying reasoning, creating insufficient understanding. “AI-Integrated QSAR Modeling” explains that simpler models are more comprehensible but capture only surface-level relationships, whereas more complex ones are more accurate but harder to trust. Modern QSAR is moving from a black box to more explainable data by using visuals and AI. Validation techniques can assess the accuracy of QSAR models. Internal validation assesses the consistency of the results by repeatedly running the algorithm, while external validation uses new datasets to evaluate its performance on compounds not used in training. Shuffling data and performing a randomization test are other ways to ensure that the predictions it makes are not random. Until QSAR can overcome this “black box” and communicate effectively, its predictive results will not be completely trusted by scientists.
Activity cliffs are another significant challenge for QSAR models. An activity cliff occurs when two structurally very similar molecules exhibit dramatically different levels of biological activity. This directly contradicts the foundational idea of QSAR that similar structures lead to similar activity, creating a major reliability problem for predictive models. When a dataset contains many activity cliffs, the model encounters contradictory signals during its training, making it difficult to learn consistent and accurate patterns. Research comparing nine QSAR models across three drug targets found that all models struggled significantly when predicting the activity of both molecules in a cliff pair from scratch. Even the descriptor type or algorithm used didn’t help. However, performance improved when the activity of one molecule was already known, suggesting that models rely heavily on reference points rather than truly understanding structural relationships, which poses a significant problem. Removing activity cliffs from training data is not a viable solution either, as it eliminates valuable structure-activity relationship information the model needs for predictions. A higher density of activity cliffs in a dataset consistently correlates with poorer overall model performance, making this a modern unsolved problem in QSAR research.
Conclusion
QSAR has evolved significantly, shifting from traditional statistics-based modeling software to an advanced program that uses machine learning and deep learning. It has become an integral part of modern drug discovery and chemical research, reducing time, cost, and resources spent on pharmaceutical testing. While early QSAR models utilized simpler methods such as linear regression, they have evolved to use support vector machines and neural networks to analyze complex molecular relationships that they could not before. These developments have enabled researchers to analyze massive libraries of molecular data much more efficiently, providing a framework for many modern-day advancements.
Despite these advances, QSAR still faces several limitations, such as overfitting, inconsistent datasets, and weak reproducibility. These limitations reduce the reliability of its predictions, creating a need for validation methods that are essential tools for ensuring the model’s credibility. Resources like QSAR DataBank have enabled more accurate data collection and greater standardization across models, providing greater reliability and reproducibility. While QSAR is still in development and has setbacks, it will continue to evolve and eventually become a powerful tool in the pharmaceutical industry and for modern biomedical research.
References
Dablander, M., Hanser, T., Lambiotte, R., & Morris, G. M. (2023, April 17). Exploring QSAR models for activity-cliff prediction. Journal of Cheminformatics. Retrieved May 24, 2026, from https://link.springer.com/article/10.1186/s13321-023-00708-w
Dablander, M., Hanser, T., Lambiotte, R., & Morris, G. M. (2023, April 17). Exploring QSAR models for activity-cliff prediction. Journal of Cheminformatics. Retrieved May 24, 2026, from https://pmc.ncbi.nlm.nih.gov/articles/PMC10107580/
Gedeck, P., Rohde, B., & Bartels, C. (2006, August 03). QSAR − How Good Is It in Practice? Comparison of Descriptor Sets on an Unbiased Cross Section of Corporate Data Sets. Journal of Chemical Information and Modeling. Retrieved May 24, 2026, from https://pubs.acs.org/doi/10.1021/ci050413p
Insilico Medicine. (2022, June). From target discovery to phase 1 initiation in under 30 months: AI-discovered and designed drug enters the clinic. Biopharma Dealmakers. Retrieved May 24, 2026, from https://www.nature.com/articles/d43747-022-00112-7
Koirala, M., Yan, L., Mohamed, Z., & DiPaola, M. (2025, September 25). AI-Integrated QSAR Modeling for Enhanced Drug Discovery: From Classical Approaches to Deep Learning and Structural Insight. International Journal of Molecular Sciences. Retrieved May 24, 2026, from https://pmc.ncbi.nlm.nih.gov/articles/PMC12525248/
Li, J., Zhao, T., Yang, Q., Du, S., & Xu, L. (2025, January 15). A review of quantitative structure-activity relationship: The development and current status of data sets, molecular descriptors and mathematical models. Chemometrics and Intelligent Laboratory Systems. Retrieved May 24, 2026, from https://www.sciencedirect.com/science/article/abs/pii/S0169743924002181
Neves, B. J., Braga, R. C., Melo-Filho, C. C., Moreira-Filho, J. T., Muratov, E. N., & Andrade, C. H. (2018, November 13). QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Frontiers in Pharmacology. Retrieved May 24, 2026, from https://pmc.ncbi.nlm.nih.gov/articles/PMC6262347/?utm_source=chatgpt.com
Niazi, S. K., & Mariam, Z. (2023, July 15). Recent Advances in Machine-Learning-Based Chemoinformatics: A Comprehensive Review. International Journal of Molecular Sciences. Retrieved May 24, 2026, from https://pmc.ncbi.nlm.nih.gov/articles/PMC10380192/
Ren, F., Aliper, A., Chen, J., Zhao, H., Rao, S., Kuppe, C., Ozerov, I. V., Zhang, M., Witte, K., Kruse, C., Aladinskiy, V., Ivanenkov, Y., Polykovskiy, D., Fu, Y., Babin, E., Qiao, J., Liang, X., Mou, Z., Wang, H., … Zhavoronkov, A. (2024, March 08). A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models. Nature Biotechnology. Retrieved May 24, 2026, from https://www.nature.com/articles/s41587-024-02143-0
Ruusmann, V., Sild, S., & Maran, U. (2014, May 14). QSAR DataBank - an approach for the digital organization and archiving of QSAR model information. Journal of Cheminformatics. Retrieved May 24, 2026, from https://pmc.ncbi.nlm.nih.gov/articles/PMC4047268/
Shahin, R., Jaafreh, S., & Azzam, Y. (2025, April 04). Tracking protein kinase targeting advances: integrating QSAR into machine learning for kinase-targeted drug discovery. Future Science OA. Retrieved May 24, 2026, from https://www.tandfonline.com/doi/full/10.1080/20565623.2025.2483631#abstract
Singh, B., Crasto, M., Ravi, K., & Singh, S. (2024, October). Pharmaceutical advances: Integrating artificial intelligence in QSAR, combinatorial and green chemistry practices. Intelligent Pharmacy. Retrieved May 24, 2026, from https://www.sciencedirect.com/science/article/pii/S2949866X24000650
Tropsha, A., Isayev, O., Varnek, A., Schneider, G., & Cherkasov, A. (2023, December 08). Integrating QSAR modelling and deep learning in drug discovery: the emergence of deep QSAR. Nature Reviews Drug Discovery. Retrieved May 24, 2026, from https://www.nature.com/articles/s41573-023-00832-0
Vasilev, B., & Atanasova, M. (2025, January 24). A (Comprehensive) Review of the Application of Quantitative Structure–Activity Relationship (QSAR) in the Prediction of New Compounds with Anti-Breast Cancer Activity. Applied Sciences. Retrieved May 24, 2026, from https://www.mdpi.com/2076-3417/15/3/1206
Veerasamy, R., Rajak, H., Jain, A., Sivadasan, S., Varghese, C. P., & Agrawal, R. K. (2011, September). Validation of QSAR Models - Strategies and Importance. International Journal of Drug Design and Discovery. Retrieved May 24, 2026, from https://www.researchgate.net/profile/Ravichandran-Veerasamy/publication/284566093_Validation_of_QSAR_Models_-_Strategies_and_Importance/links/5ca57788458515f78522300e/Validation-of-QSAR-Models-Strategies-and-Importance.pdf
Zhang, F., Wang, Z., Peijnenburg, W. J., & Vijver, M. G. (2023, July). Machine learning-driven QSAR models for predicting the mixture toxicity of nanoparticles. Environment International. Retrieved May 24, 2026, from https://www.sciencedirect.com/science/article/pii/S0160412023002982

Thank you to Rishi Shah for creating and maintaining the website.